![Development of our tool was driven by real molecular-biology issues poisoning 454 and IonTorrent data]()
There is no other 454 adapter trimming tool looking for real issues.
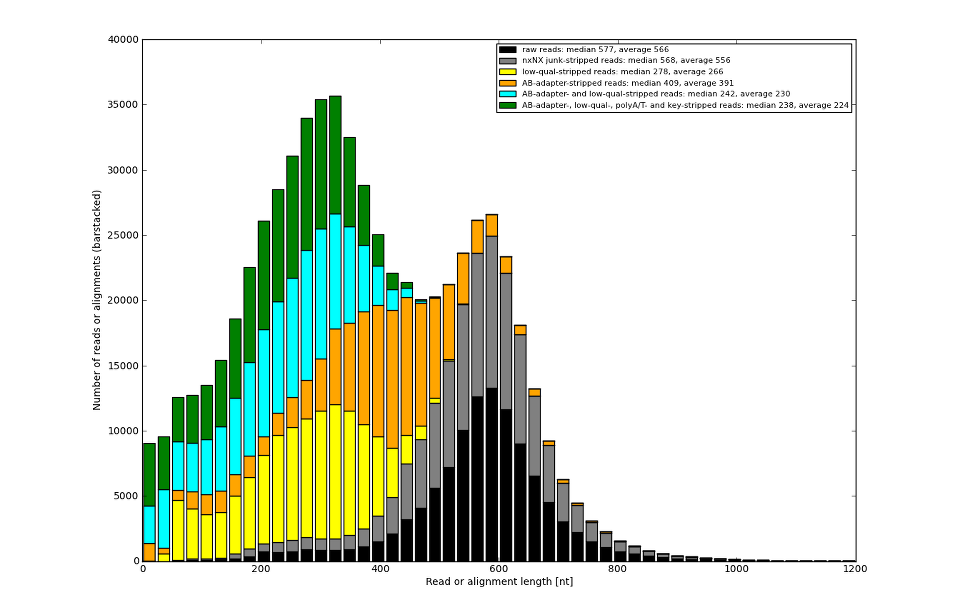
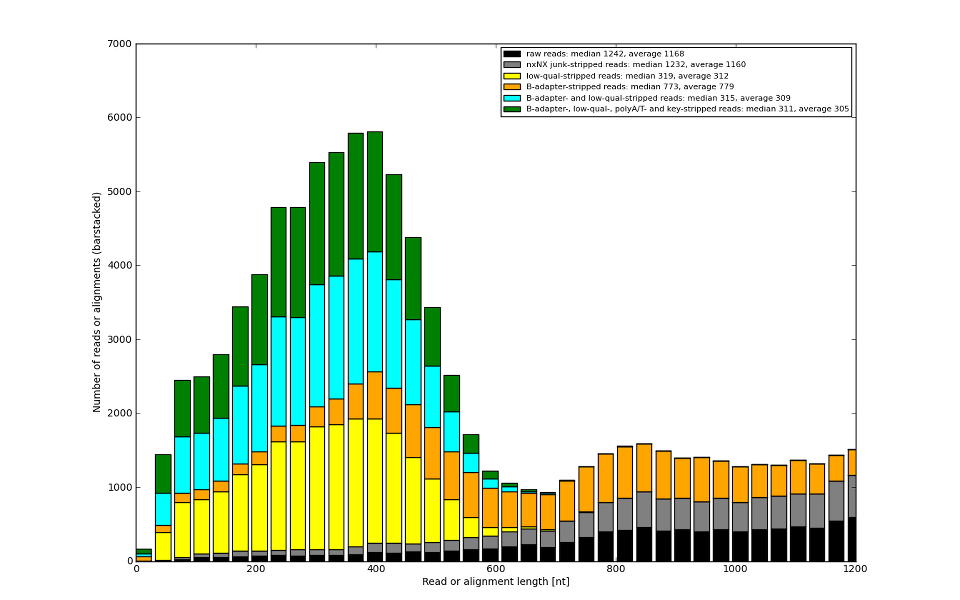
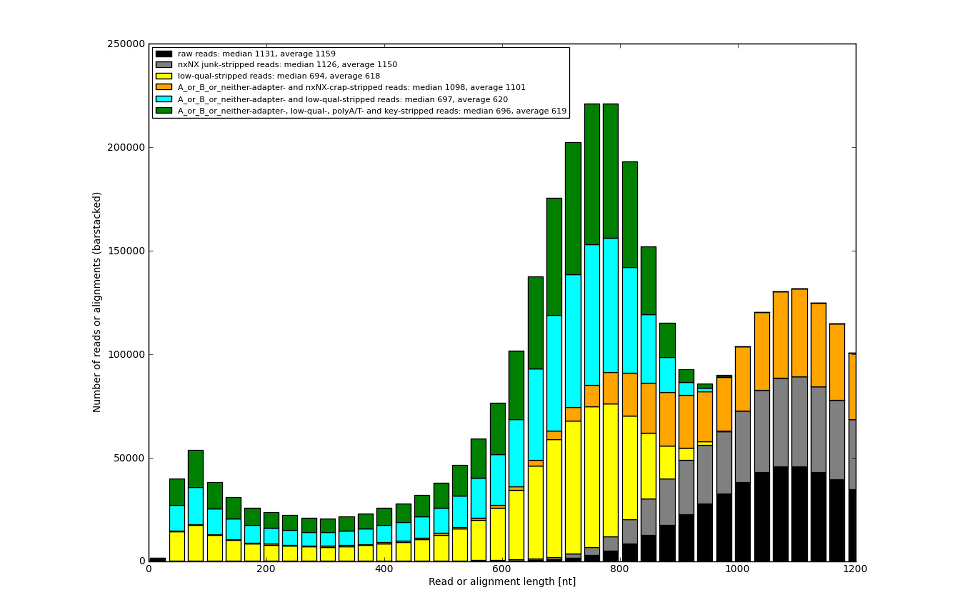
We can at least rescue part of the deemed low-qual sequence to make the final sequence longer.
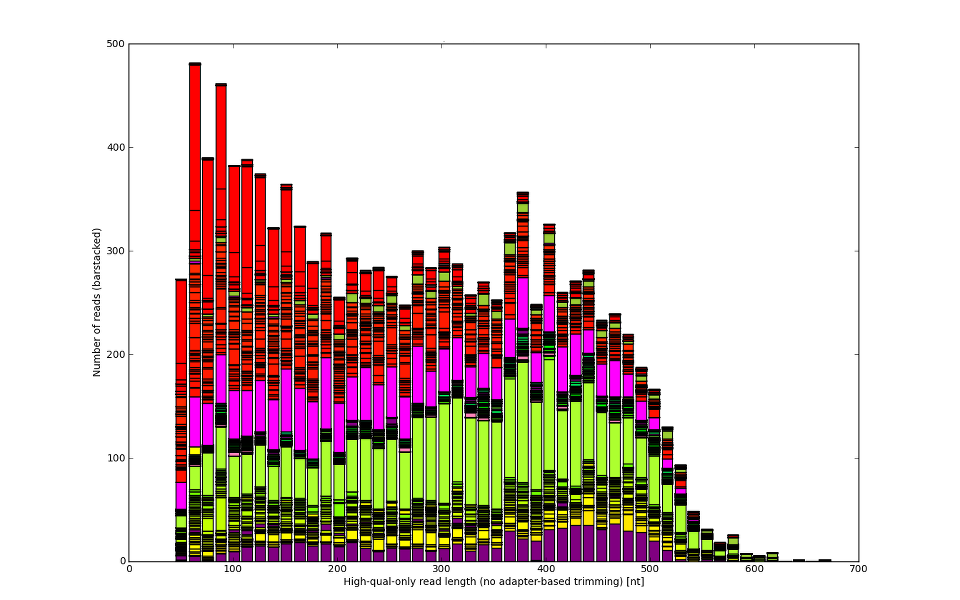
Not always is the sequencer or vendor to be blamed.
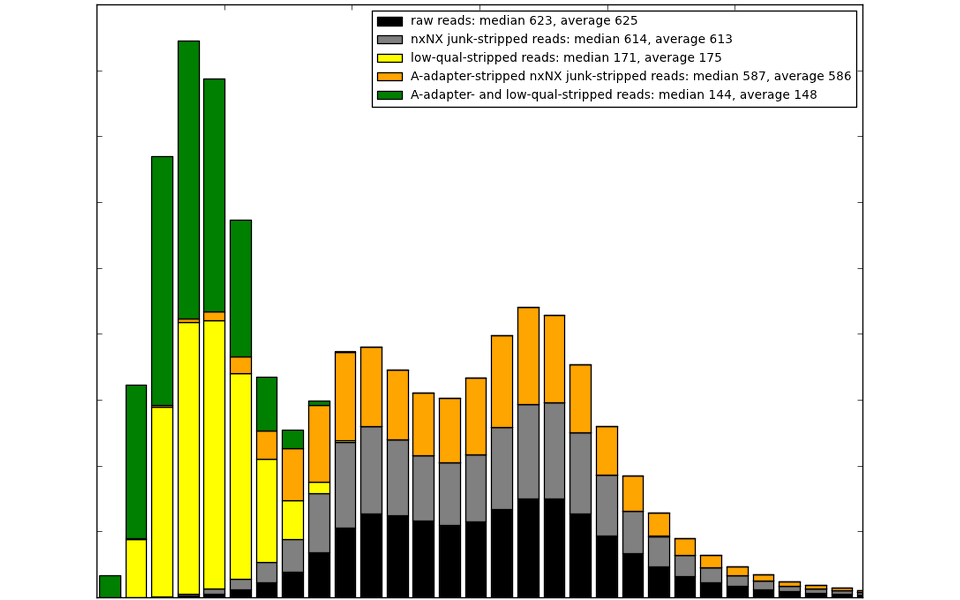
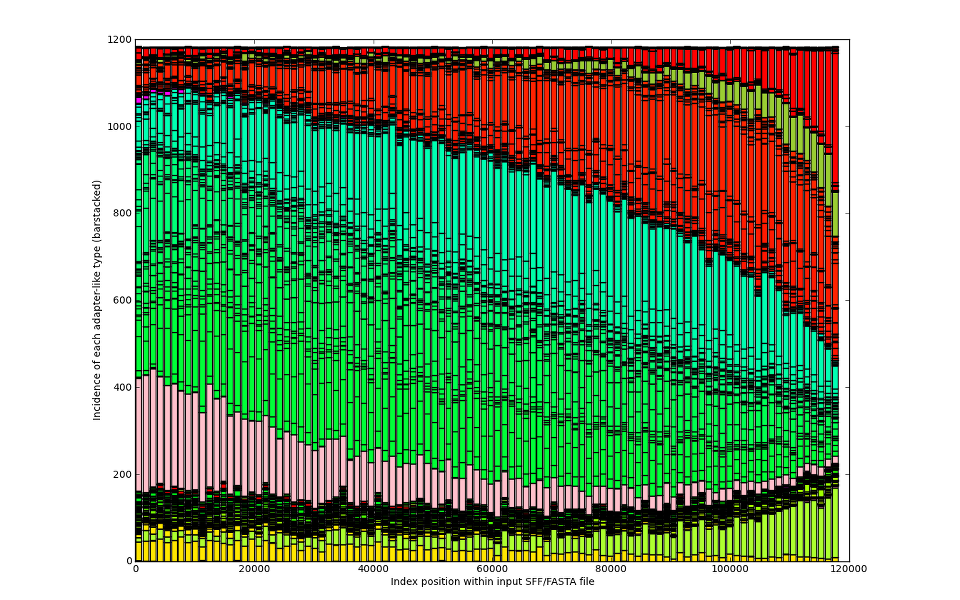
We can remove the offending adapters/MIDs/artefacts from your reads.
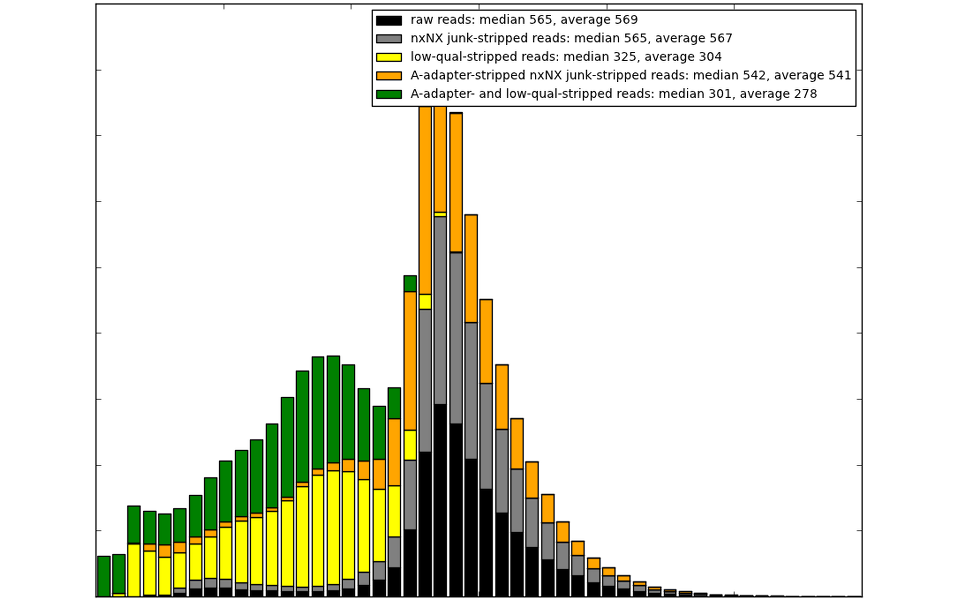
Unremoved adapters prevent contig joining and scaffolding.
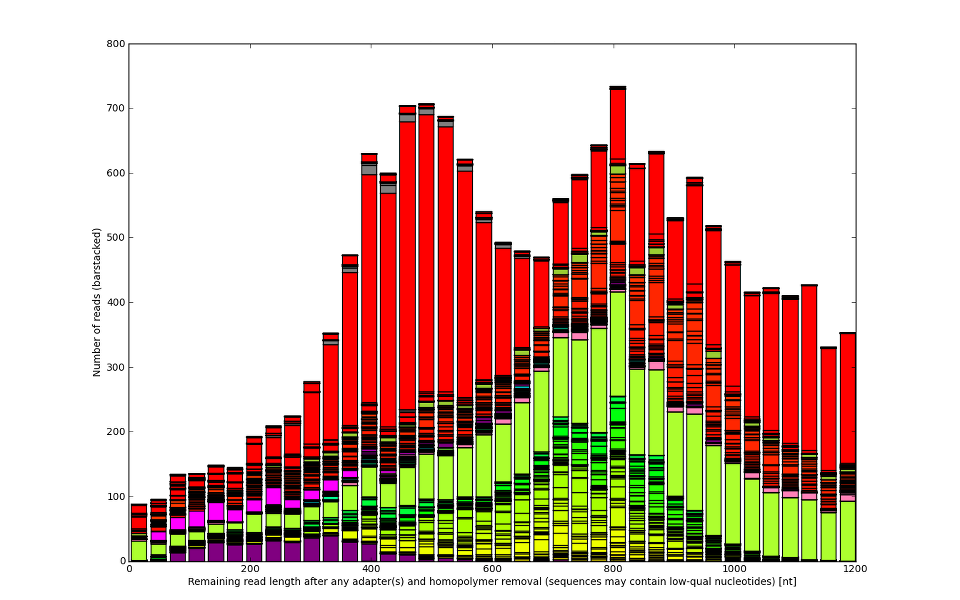
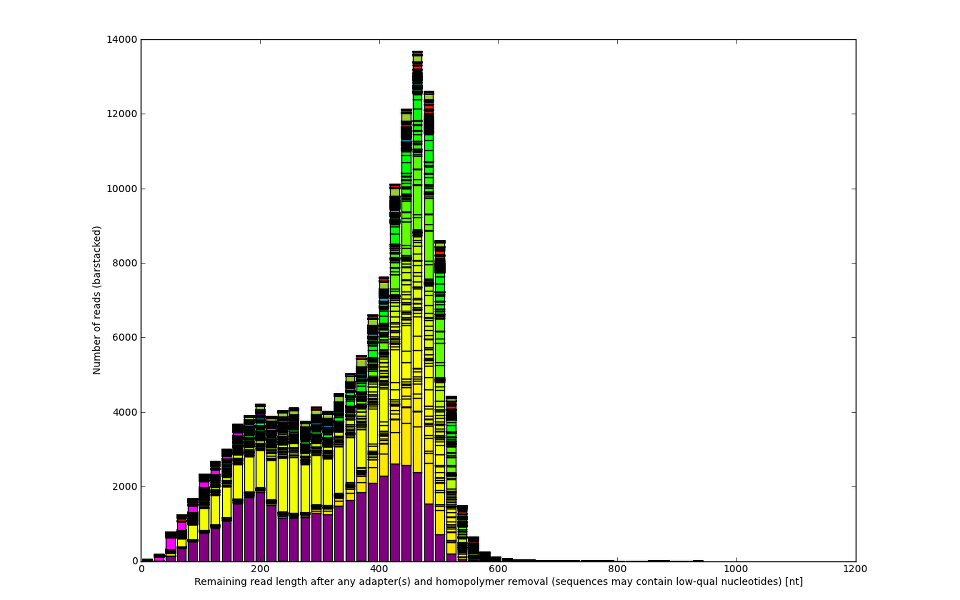
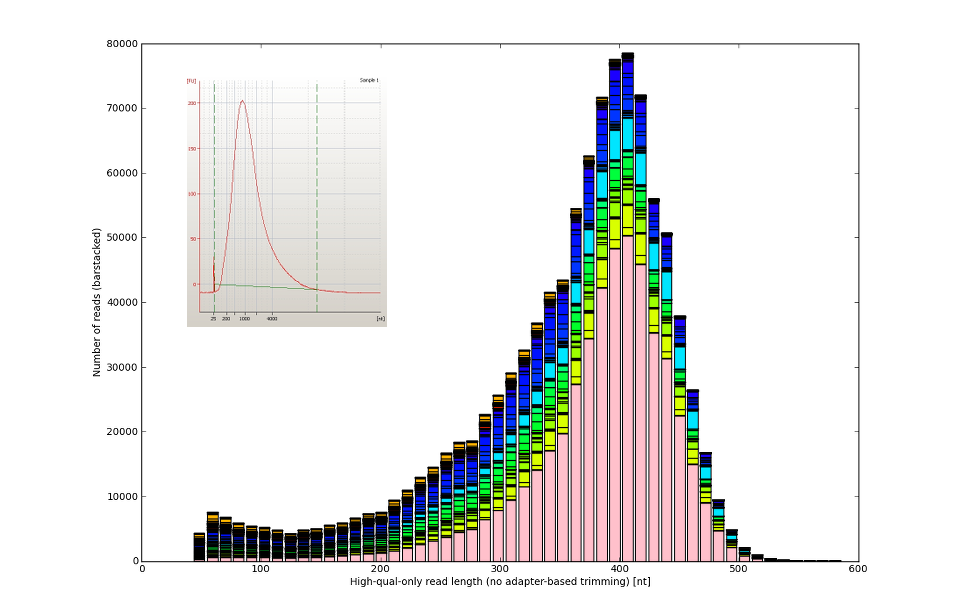
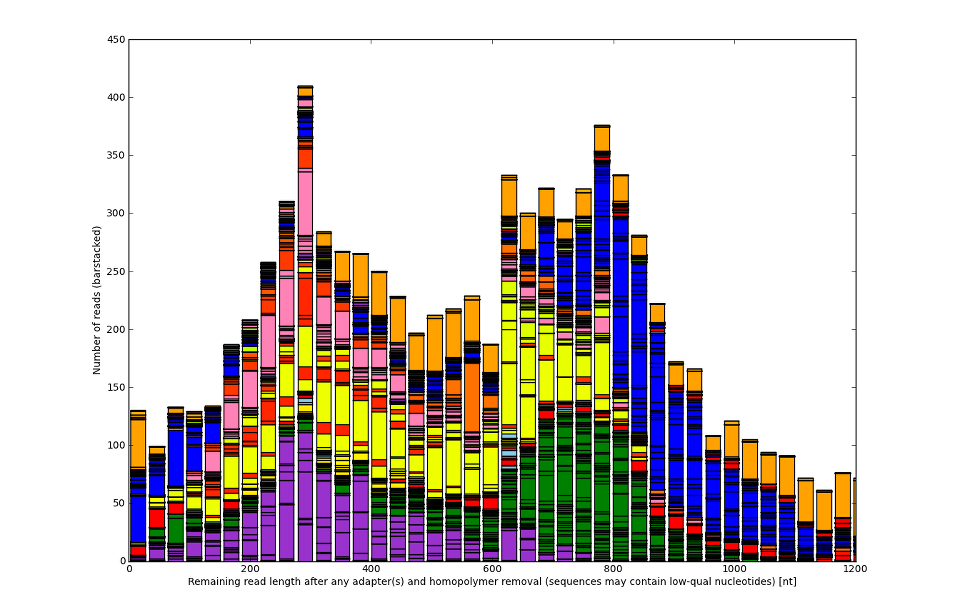
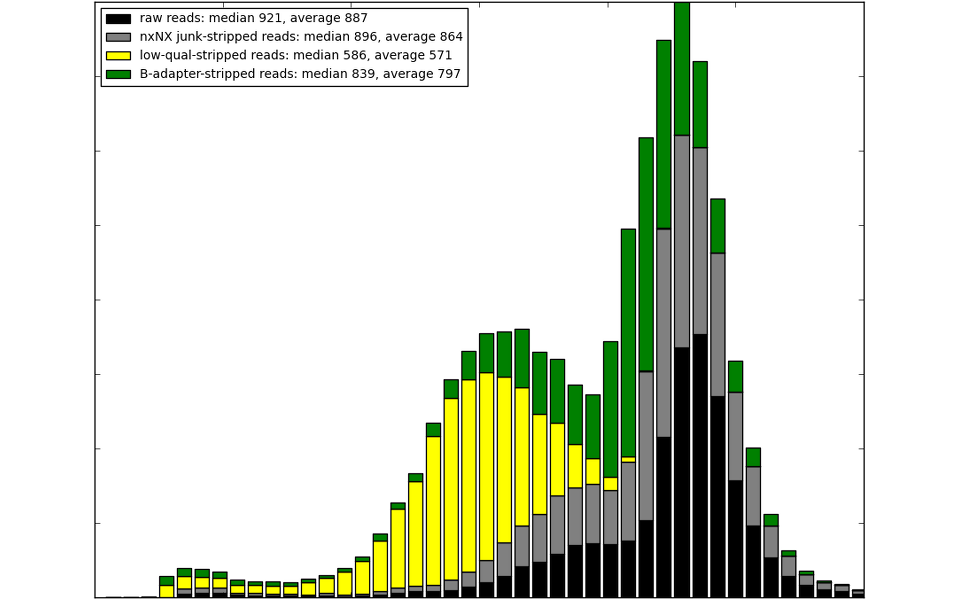
Sometimes you could gain even 200nt of perfectly usable sequence.
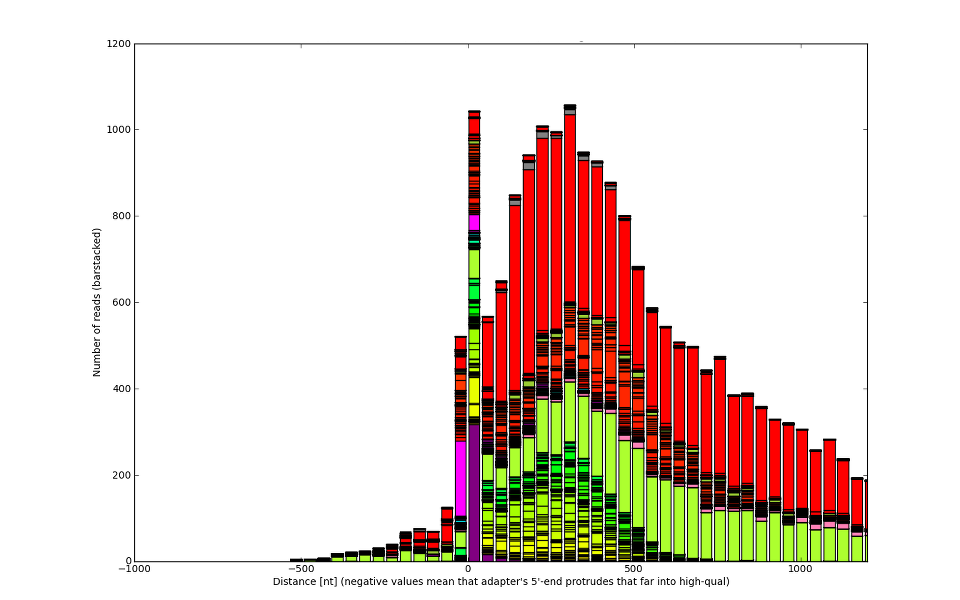
In overall, minimize assembly artefacts, make the assembly feasible and correct.
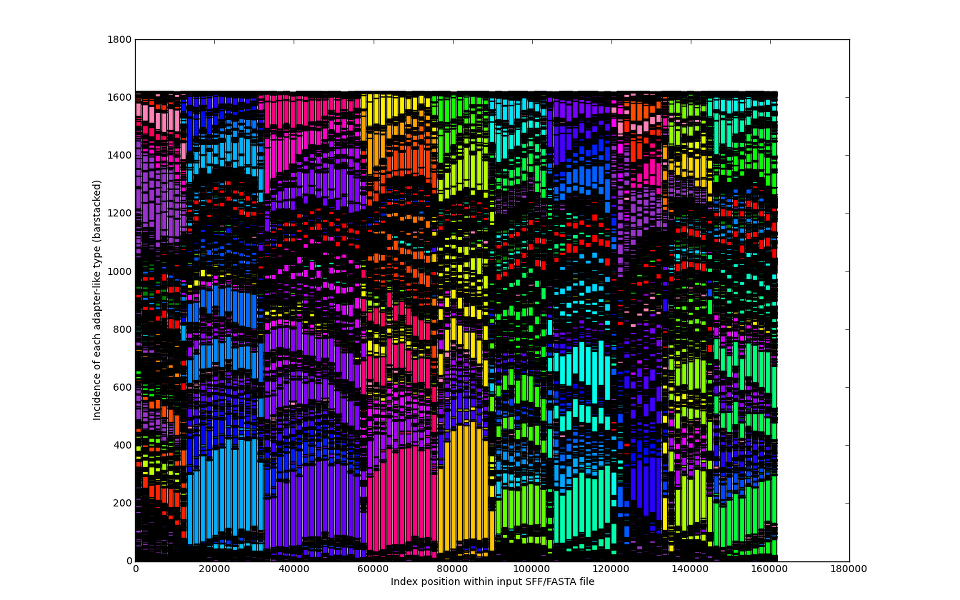
We learned that from never-ending analyses of >1800 independent sample datasets
Sometimes we can tell you based on the data what you did wrong.
We can compare the results and tell you which was your best trial.
![We fix what others overlooked in the past.]()
We are fixing bugs still present in software from Roche and IonTorrent.
We Have Been Featured In The Following Publications